Forensic science tends to be a slow-moving field, but every so often new technology helps push forensic testing forward. Massively parallel sequencing (MPS) is one such technology with substantial potential benefit for forensic DNA testing. Through a technology transfer partnership, Ohio BCI and the Battelle Memorial Institute (Columbus, Ohio) have implemented MPS-based mitochondrial DNA (mtDNA) testing into BCI’s Missing/Unidentified Persons casework. Performing MPS-based mtDNA testing in-house allows Ohio BCI to offer a complete and improved testing portfolio to the many law enforcement agencies it serves.

Written by: Adam Garver, Ohio Bureau of Criminal Investigation (BCI)
As part of validations of two MPS-based testing kits, the BCI and Battelle team have demonstrated proficiency with multiple MPS workflows. The summaries of those validations were submitted to the FBI, which subsequently approved data from the kits to be uploaded to the Combined DNA Index System (CODIS). To make integration as seamless as possible, the BCI-Battelle team tested the compatibility of time-saving crude extracts and direct amplification with MPS workflows. The team evaluated three MPS products for use with crude extracts from reference samples: a prototype version of the Promega PowerSeqTM Auto/Y System, the Illumina ForenSeqTM kit with DNA Primer Mix A (DPMA), and the Promega PowerSeqTM CRM Nested System, Custom.
For the study, forty-nine1.2 mm punches from reference saliva samples on Bode Buccal DNA Collectors made of cotton filter paper (Bode Cellmark Forensics, Lorton, VA, USA) were punched with a BSD600 Duet (BSD, Queensland, Australia) into 10 μL PunchSolutionTM (Promega Corp). The samples were incubated at 70 ̊C for 30 minutes, and then 45 μL 1X Tris-EDTA (TE) DNA Suspension Buffer (Teknova, Inc. Hollister, CA) was added and pipette mixed 10 times. Then either 5µl was taken directly for amplification, or 10µl of a diluted amount of crude extract was added to the amplification reaction.
The PowerSeq™ Auto/Y System data were compared to Sanger based capillary electrophoresis (CE) data generated using previously validated methods. Forty-eight of the 49 samples resulted in profiles concordant with CE data at all typed autosomal STR (aSTR) loci. The one exception was observed in a single reference sample that exhibited low coverage and imbalance at TPOX and a single instance of allele drop out at D7S820. Twenty male samples had YFiler™ Plus (Thermo Fisher Scientific) data available for comparison, and all produced full Y-STR profiles fully concordant with the YFiler™ Plus results. Intralocus balance was evaluated as read count ratio (RCR), and direct amplification of reference samples produced well-balanced RCRs (averaging approximately 70%) across all aSTR loci (Figure 1).
The ForenSeq™ kit was used to type 30 reference samples with DNA Primer Mix A. The average RCR ranged from 28.3% at D22S1045 to 90.8% at D4S2408 (Figure 1). Twenty-nine of the 30 samples analyzed with the default Universal Analysis Software (UAS; Illumina) parameters, were concordant with CE. One sample exhibited apparent drop-out at TPOX due to imbalance; the allele call was user-deleted due to an RCR under 14% of its’ sister allele and being located in an n+1 stutter position.
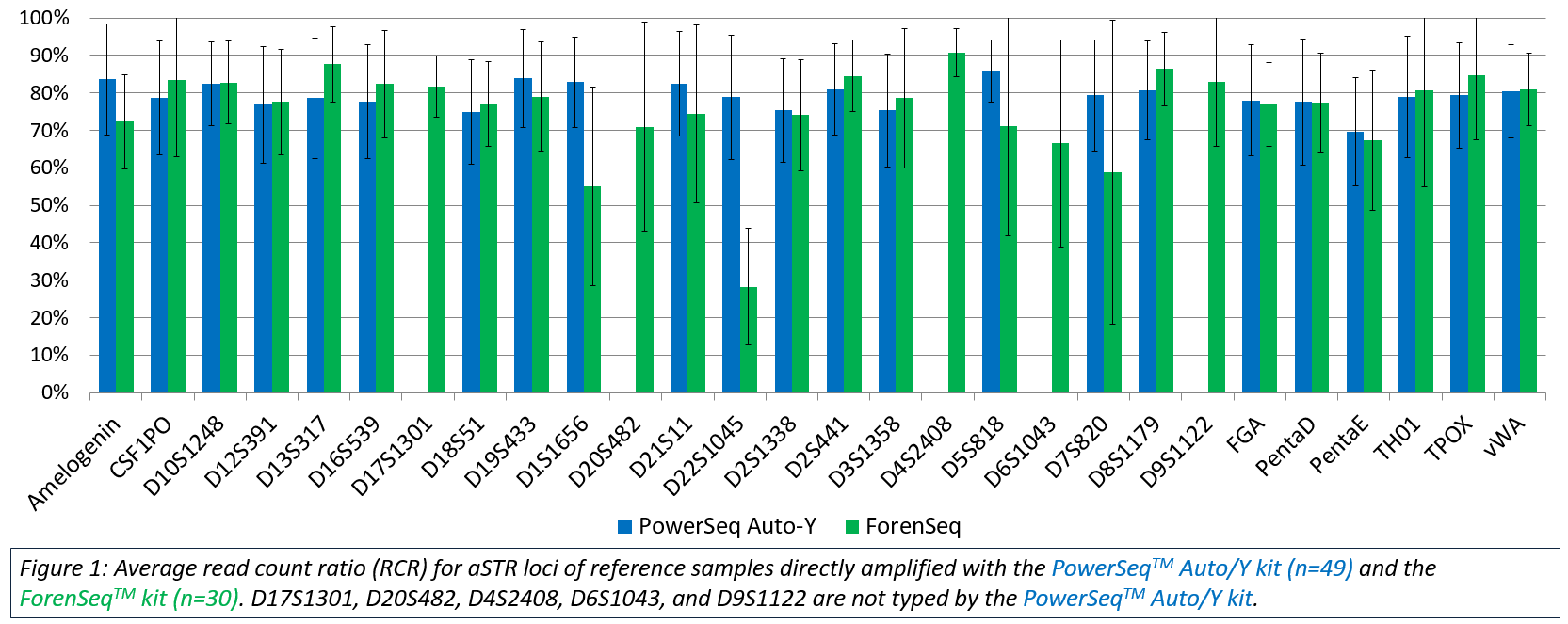
Thirty reference samples were amplified with both the PowerSeq™ Auto/Y System and the ForenSeq™ kit, and length-based allele data for all 30 samples were concordant at every locus where data were produced in both kits. In addition, the allele sequence data were compared and found to be 100% concordant. As observed with this testing, accurate genotypes and sequence data were produced from directly amplified reference samples regardless of the MPS kit used.
The PowerSeq™ CRM Nested System was tested with 15 reference samples with inputs adjusted to a target of 1,000 mitochondrial copies for amplification. Average coverage across the control region was variable (Figure 2); however, the average coverage per observed variant was greater than 10,000 reads.

Crude extracts were sent to the Emerging Technologies Section, Armed Forces Medical Examiner System’s Armed Forces DNA Identification Laboratory (AFDIL) for mtDNA control region analysis using orthogonal MPS workflows. Thirteen of the 15 samples tested at BCI generated high coverage in the control region and produced results fully concordant with AFDIL variant calls (data not shown). One sample was removed from the comparison due to low coverage across most of the control region, and one sample was concordant with AFDIL results at variants with coverage, but exhibited some regions of low coverage; therefore, two variants identified by AFDIL were not reported.
This testing demonstrates that crude extracts from single-source reference samples routinely used for direct amplification in CE analyses may also be utilized for MPS analysis. The use of crude extracts from reference samples in MPS workflows allows for a decrease of several hours processing time due to elimination of sample extraction. This time savings, as well as the ability to multiplex different marker types into one reaction, allow for the use of MPS as a viable alternative to CE-based testing in forensic laboratories.
Over the coming months, the Ohio BCI and Battelle team will continue to gain experience with MPS as this technology is applied to missing/unidentified persons casework and showcase the value of MPS to forensic DNA analysis. To adept the workflow to high-throughput conditions, bone sample extraction and post-amplification steps are being automated.
Acknowledgments: Ohio BCI would like to thank Doug Storts and Spencer Hermanson at Promega as well as Melissa Kotkin, Meghan Didier, and Cydne Holt at Verogen for providing reagents, technical guidance, and data analysis support. The authors would also like to thank Kimberly Andreaggi, Joseph Ring, Cassandra Rachel Taylor, and Charla Marshall at AFDIL for providing additional mtDNA analysis and data support. And finally, the authors thank Michael Brandhagen and Mitchell Holland for their technical assistance and guidance.
WOULD YOU LIKE TO SEE MORE ARTICLES LIKE THIS? SUBSCRIBE TO THE ISHI BLOG BELOW!
SUBSCRIBE NOW!