Today’s guest blog is written by Jianye Ge, University of North Texas Health Science Center
Deconvoluting mixture samples is one of the most challenging problems in forensics. Efforts have been made to provide solutions regarding mixture interpretation. The probabilistic interpretation of the Short Tandem Repeat (STR) profiles can increase the number of complex mixtures that can be analyzed. A portion of complex mixture profiles, particularly for mixtures with a high number of contributors, are still being deemed uninterpretable. Novel forensic markers, such as Single Nucleotide Polymorphisms (SNPs), Insertion-Deletion (Indels), and microhaplotypes, also have been proposed to allow for better mixture deconvolution. However, these markers either have lower discrimination power compared with STRs and are not compatible with CODIS. The short-read sequencing (SRS) technologies can facilitate mixture interpretation by identifying the intra-allelic variations within STRs. Unfortunately, the limited sizes of STR markers and the short-reads limit the number of alleles that can be attained per STR.
I am always exploring new technologies that could help with forensic problems. Long-read sequencing is one technology that has become more mature recently and has been successfully used in many other biomedical research and applications. The long-read sequencing enables the detection and phasing of a large allele/region missed by short-read sequencing, which fits very well with the need for mixture interpretation.

The latest long-read sequencing (LRS) technologies (e.g., Pacific Biosciences or PacBio) can overcome this limit in some samples and sequence larger DNA fragments (including STRs, SNPs, and Indels) with definitive phasing. Thus, more information could be kept and combined to improve the following mixture deconvolution.
Many people in the Center for Human Identification contributed to this work, including Bruce Budowle, Xuewen Wang, Muyi Liu, Jonathan King, Melissa Muenzler, Sammed Mandape. Also, NIJ funded this project to develop the whole sequencing and bioinformatics workflow (15PNIJ-21-GG-04159-RESS)
We developed a novel forensic marker, macrohaplotype, which combines CODIS STR and flanking variants in an extended fashion. Figure 1 shows the general design of a macrohaplotype, in which a CODIS STR is centered in the amplicon (although it can sit anywhere in the amplicon), together with non-CODIS STRs and SNVs in the flanking regions. A macrohaplotype is a haplotype with the alleles of all markers on the same paternal or maternal chromosome, which can be captured. By anchoring on forensic standard STRs, there are data that would be compatible with current databases and additional data to better serve to deconvolve mixtures.
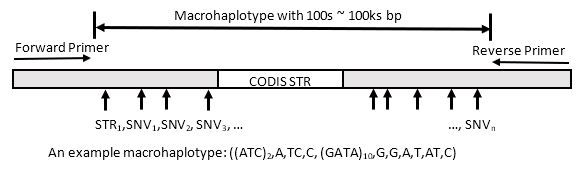
Following this novel idea, a panel of 20 macrohaplotypes with maximum discriminating power of ~8k bp in length has been designed based on the 1000 genomes project data. The primers of these macrohaplotypes have been designed. A PacBio SMRT sequencing workflow and a bioinformatics pipeline are under development using known genome cell line samples.
The statistical evaluation based on the 1000 genomes project data showed that these macrohaplotypes substantially outperform CODIS STRs for mixture interpretation, particularly for mixtures with a high number of contributors. In terms of Random Match Probability, one macrohaplotype is equivalent to 3~4 CODIS STRs. In terms of Probability of Exclusion, one macrohaplotype is equivalent to 3~8 CODIS STRs, depending on the Number of Contributors (NOC). More importantly, macrohaplotypes can much better estimate the NOC of the mixtures, particularly for mixtures with high NOC, as much less overlap between the alleles from different contributors would be observed with macrohaplotypes.
We are currently developing a PacBio SMRT sequencing workflow to generate DNA reads and a bioinformatics pipeline to call the macrohaplotypes from the DNA reads. The initial results have shown that the macrohaplotype could be successfully detected from low-input samples.
Based on the experiences at the Center of Human Identification, only 10~15% of the mixture samples are highly degraded. Thus, the majority of the mixture samples could be sequenced with long-read sequencing technology to generate more meaningful information for the subsequent data interpretation.
This project would fundamentally enhance the ability to more effectively interpret mixtures. The results from this study will expand the number of samples (and hence cases) where forensic biological evidence can provide valuable investigative leads, especially for highly complex mixtures and thus assist in solving more cases. I am also working on another solution to tackle the mixture challenge by single cell profiling, which will be the new paradigm for mixture interpretation.
WOULD YOU LIKE TO SEE MORE ARTICLES LIKE THIS? SUBSCRIBE TO THE ISHI BLOG BELOW!
SUBSCRIBE NOW!